
Fig. 1. Electron micrograph of a single, complete Citrus tristeza virus particle. Sap from infected a citrus leaf was spread directly on the EM grid. The virus particle was negatively stained with uranyl acetate (Z. Xiong).
Citrus tristeza virus (CTV) is a member of the genus Closterovirus within the family Closteroviridae. It is the most important and destructive virus of citrus. CTV virions (Fig. 1) are flexuous rods, 2000 nm in length and 12 nm in diameter, consisting of one single-stranded, (+)-sense RNA genome encapsidated by two species of coat proteins (97% CP and 3% CPm).
|
Fig. 1. Electron micrograph of a single, complete Citrus tristeza virus particle. Sap from infected a citrus leaf was spread directly on the EM grid. The virus particle was negatively stained with uranyl acetate (Z. Xiong). |
The 19.2 to 19.3 kb CTV genome, one of the largest in RNA viruses, contains 12 open reading frames. The 5’ half of the genome is translated directly to a large, frameshifting protein that contains motifs characteristic of RNA-dependent RNA polymerase, helicase, methyltransferase, and proteases. This protein is required for CTV replication and is most likely proteolytically cleaved into smaller, functional proteins. The 3’ half encodes ten proteins expressed from ten 3’ co-terminal subgenomic RNAs. These proteins include viral structural proteins and proteins interacting with host plants.
CTV is perhaps one of the most diverse viruses with numerous, disparate strains, each inducing different types and degrees of disease symptoms on different citrus species and varieties. In nature, CTV often exists as a complex, comprising multiple strains or genotypes due to the longevity of citrus trees and their vegetative propagation by budwood. Continual vertical transmission coupled to repeated horizontal transmission mediated by aphids throughout the history of citrus cultivation has led to the complexity of the CTV population increasing over hundreds of years, resulting in the co-existence of multiple CTV genotypes in a single host. The presence within a host of multiple replicating CTV genotypes and the relatively long periods of co-replication create opportunities for recombination between the genotypes, leading to extensive viral diversity.
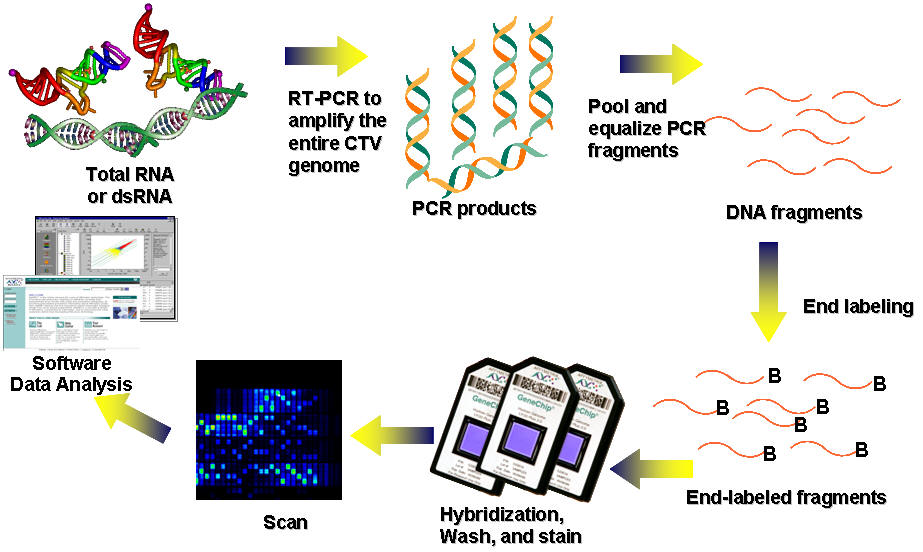
Fig. 1. Simplified work flow of resequencing analysis of Citrus tristeza virus.
To rapidly sequence a large number of CTV genomes and to study the diversity of the CTV complex at the sequence level, we designed and validated an Affymetrix resequencing microarray that queries entire genomes of multiple CTV genotypes. Full genomic sequences of representative CTV isolates were selected for microarray tiling because of the tiling capacity of the microarray (Table 1). The selection was based on the phylogenetic analysis of fully sequenced CTV genomes available at the time of the microarray design. Genomes of the remaining CTV isolates were then compared to that of the tiled and the most closely related CTV genome to identify unique sequences of the genomes for additional microarray tiling. The complete tiling comprises four full-length CTV genomes, one partial genome, and unique sequences from other CTV isolates, representing a genetic diversity equivalent to that contained in ten complete CTV genomes.
Table 1. CTV sequences tiled on the resequencing microarray
|
CTV strains/isolates
|
No. of Bases
|
|
T30
|
19,259
|
|
T36
|
19,293
|
|
VT
|
19,226
|
|
T3
|
19,253
|
|
T68-1
|
13,585
|
|
SY568 unique
sequence
(5’ VT + 3’ T30)
|
8,090
|
|
H33 unique sequence (VT)
|
8,391
|
|
NUagA unique sequence (VT)
|
9,991
|
|
T385 unique sequence (T30)
|
127
|
|
Qaha unique sequence (T36)
|
2,298
|
|
Total nucleotides
|
117,088
|
|
Internal control nucleotides
|
807
|
|
Total Probes
|
943,160
|
CTV genomic DNA required for resequencing analysis was obtained by long-range reverse transcription-polymerase chain reaction (RT-PCR), using four sets of universal primers (Table 2). Each set of primers consisted of an RT primer and a pair of PCR primers, and were designed using sequences highly conserved in all known CTV genomes. Together, the four sets of primers were capable of amplifying entire genomes of all known CTV isolates as four DNA fragments ranging from 4.5 to 5.5 kb (Fig. 2).

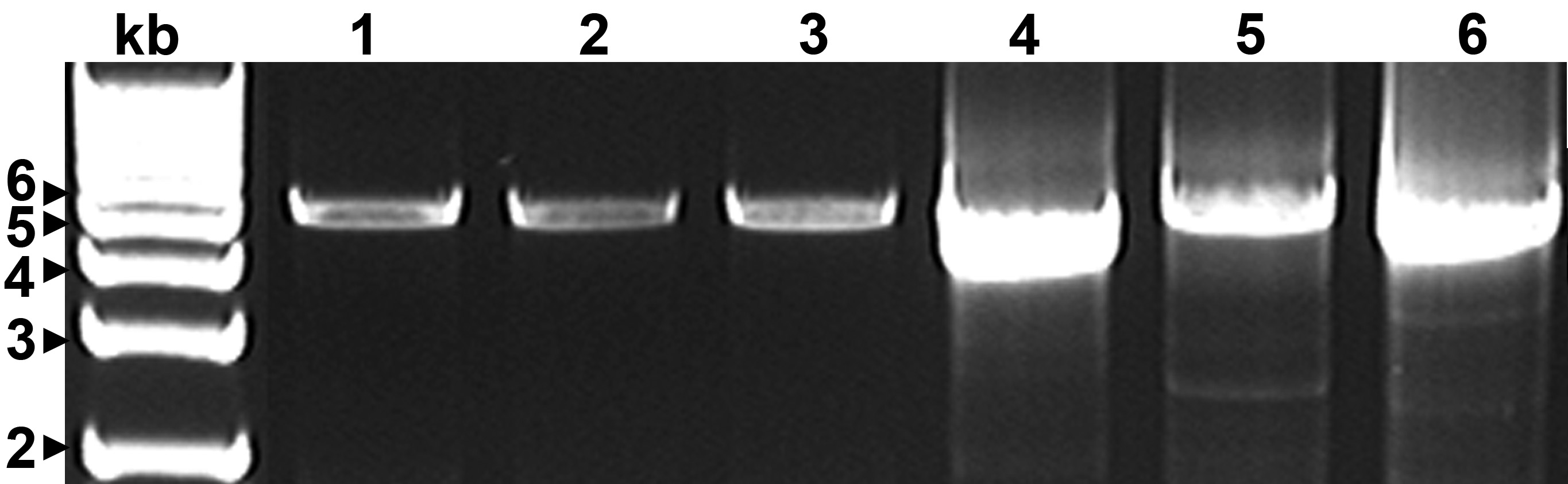
Fig. 2. DNA fragments representing entire CTV genomes amplified by RT-PCR from FS2-2. 1-3, DNA amplified by the 3’ primer CTV5427R and three 5’ primers: CTV5endFT36, CTV5endFVT, and FT30CTV5end, respectively; 4, DNA amplified by primers CTV5403F and CTV09997R; 5, DNA amplified by primers CTV09262F and CTV14630R; and 6, DNA amplified by primers CTV14469F and CTV19395R. kb, 1 kb DNA ladder.
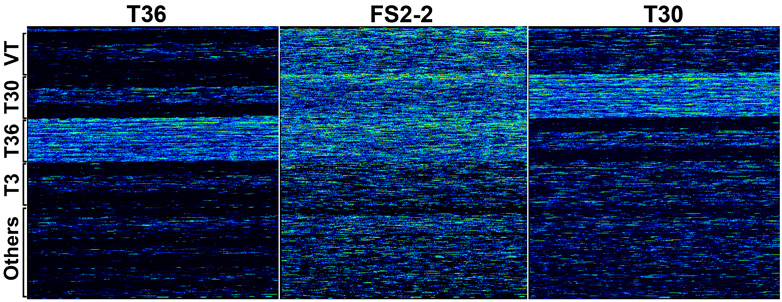
Fig. 3. Images of hybridized resequence microarrays. Affymetrix CTV Microarray chips were hybridized with target DNA from T36 and T30 strains and an unknown sample, FS2-2. Warm colors and cool colors represent higher and lower hybridization intensities, respectively. Locations of tiled CTV genomes on the microarray are indicated to the left. Single genome blocks are hybridized with T36 and T30 target DNA while multiple genome blocks are hybridized with FS2-2 target DNA . Three CTV genotypes similar to T30, T36, and VT were identified from the resequencing analysis
The massively parallel, pyrosequencing-based 454 sequencing has also been applied to CTV genomic analyses successfully. 454 sequencing is extremely powerful, with a single instrumental run producing sequences of 100 megabases or more. This tool is ideal for population genomics of viruses. To demonstrate the feasibility of using 454 sequencing for a genome-wide, concurrent sequencing analysis of multiple CTV strains, a 1/16-region 454 sequencing run was performed on a field CTV isolate containing three distinctive strains.
RT-PCR amplified CTV genomic DNA from FS2-2 were used as template DNA for the 454 sequencing in the Genome Sequencer FLX. Statistics of the sequencing run are listed in Table 3. Sequencing reads were assembled to three separate, full-length CTV genomic contigs with 27X to 43X coverage (Fig. 4). Analysis of the full-length CTV genomes suggest that they are closely related to the reported genomes of the CTV strains T30, T36, and VT. Additionally, a preliminary analysis found a large number of recombinant CTV sequences and defective CTV sequences. The recombinant sequences consist of sequences from two or more CTV strains joined together while the defective sequences comprise sequences joined from different regions of the same CTV genome. A genome-wide recombination map for each of the CTV strains in FS2-2 was constructed using these recombinant and defective sequences (Fig. 5. These maps revealed a systematic and unprecedented scale of recombination between co-infecting strains in a single host. The recombination activities occurred across the genome, with most active recombination occurring at the 3’ half of the CTV genome.
Together, the resequencing and 454 sequencing analyses provide an extremely high resolution portrait of the complexity and the evolutionary dynamics of CTV infections that has implications in using cross protection as a tristeza management strategy and in understanding emergence of new CTV strains.

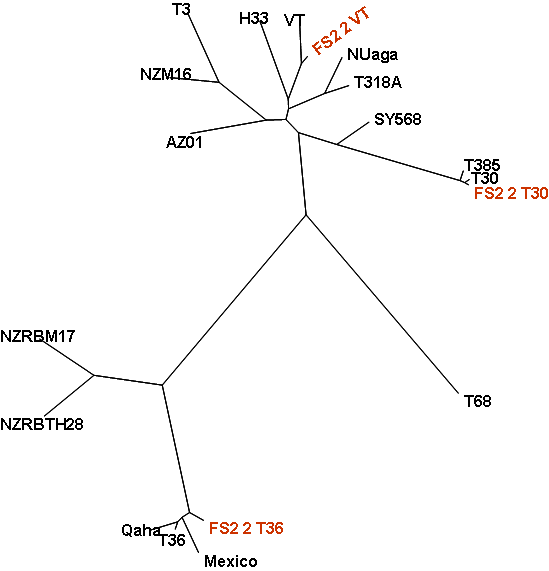
Fig. 4. A phylogenetic tree of CTV depicting the relationship of three CTV genomes (highlighted) assembled from 454 sequencing analysis of FS2-2 to other CTV genomes
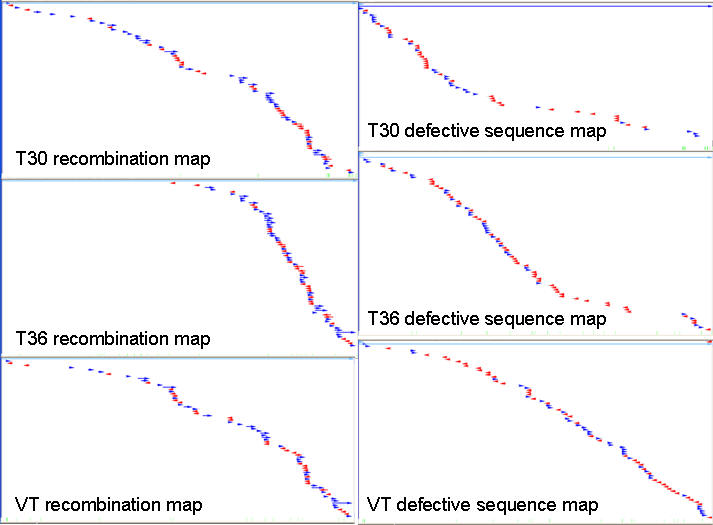
Fig. 5. CTV recombinant and defective sequence maps generated from isolate FS2-2. The line at the top of each box represents the CTV genome from the 5’ end to the 3’ end. Each arrow denotes the location of a recombinant or defective sequence aligned to the CTV genome. Blue arrows indicate sequences in the sense orientation while red arrows indicate sequences in the antisense orientation.